Abstract 1788: Mechanism of resistance to c-Met tyrosine kinase inhibitors in human melanoma
http://cancerres.aacrjournals.or ... etingAbstracts/1788
The over-expression of c-Met and activation by its ligand, hepatocyte growth factor (HGF), are known to cause significant tumor development and metastasis in human melanoma. Hence, c-Met is an attractive target for molecular therapeutic inhibition. To assess c-Met inhibition, we studied the effects of c-Met tyrosine kinase inhibitors (TKIs) in RU melanoma cells and found that JNJ38877605 and SU11274 (c-Met TKIs) inhibited cell growth in RU cells with an IC50 of 0.5 and 1µM, respectively. We then tested the therapeutic effects of JNJ38877605 in vivo. Five million RU melanoma cells were injected subcutaneously into the hind flanks of nude mice. Tumors were allowed to develop for a week after which daily oral treatments of 20 mg/kg JNJ38877605 or vehicle were given. We found that JNJ38877605 decreased the size of RU tumor xenografts in nude mice by 6 fold compared to mice treated with a vehicle control. Unfortunately, while c-Met inhibitors have been successful in clinical trials, acquired resistance to c-Met TKIs (ARQ197) and antibodies (MetMab) has been observed in cancer patients, resulting in tumor recurrence or lack of tumor CR. To determine the mechanism of c-Met TKI resistance, two melanoma cell line models, MU and RU, were exposed to increasing concentrations of SU11274. They proliferated in 7-10 fold higher concentrations of SU11274, and were cross resistant to 12 fold higher concentrations of JNJ38877605. After generating resistant cells, we measured HGF production and the expression levels of p-c-Met (Y1003) and p-c-Met (Y1234/1235) by immunoblotting to compare parental and resistant cells. We found that MU resistant cells exhibited upregulation of p-c-Met (Y1003) (6 fold) and p-c-met (Y1234/1235) (4 fold) compared to parental cells after treatment with HGF. Additionally, p-c-Met (Y1234/1235) was stable for only 30 min in parental cells compared to 60 min in resistant cells, where it was upregulated by 3-fold. This might be due to defects in c-Met ubiquitination resulting in its being recycled back or stabilized onto the membrane. Interestingly, we found that RU parental cells secreted 2 fold more HGF than the resistant cells. This indicates that resistant RU cells may not be as dependent on HGF induced activation in comparison to parental cells. In MU and RU resistant cells, we also found upregulation of p-mTOR (2 and 8 fold, respectively). Upregulation of phospho-pS6kinase (2 fold) and p-4E-BP1 (4 fold), downstream targets of p-mTOR, were also seen in MU cells. MU resistant cells also expressed increases in active β-catenin (protein downstream of Wnt signaling pathway) by 5 fold under no HGF stimulation and 2 fold after 60 min of HGF treatment. Our data indicates that the oral c-Met TKI, JNJ38877605, could be a promising therapeutic option for treating HGF producing melanoma tumors, but its effects could be further enhanced by the addition of mTOR and/or Wnt inhibitors to counteract acquired c-Met TKI resistance.
|
 肺癌历经手术-复发-脑转移,如今母亲
讲述者:一只阿狸整理者:pear
熟悉我家的朋友都知道,我母亲虽然是IB期肺癌,但是很
肺癌历经手术-复发-脑转移,如今母亲
讲述者:一只阿狸整理者:pear
熟悉我家的朋友都知道,我母亲虽然是IB期肺癌,但是很
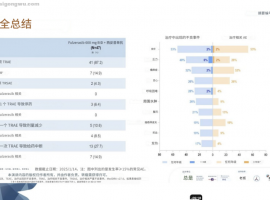 肺癌新疗法来袭!客观缓解率80%,双
作者:seacat
氟泽雷塞(商品名:达伯特®)是劲方医药开发的KRAS G12C 小分子抑制剂
肺癌新疗法来袭!客观缓解率80%,双
作者:seacat
氟泽雷塞(商品名:达伯特®)是劲方医药开发的KRAS G12C 小分子抑制剂
 医患零距离:共话ALK患者手术那些事
作者:小莱
非小细胞肺癌(NSCLC)作为肺癌的主要类型,占据了肺癌总数的85%。尽管在
医患零距离:共话ALK患者手术那些事
作者:小莱
非小细胞肺癌(NSCLC)作为肺癌的主要类型,占据了肺癌总数的85%。尽管在
 急急急,求方案,希望得到大家的帮助
群里的老师们,请问如果肾转移会左侧腰以上痛吗?已经痛了有半个多月。一开始痛的时候
急急急,求方案,希望得到大家的帮助
群里的老师们,请问如果肾转移会左侧腰以上痛吗?已经痛了有半个多月。一开始痛的时候
 与myc结合亲和力高的已上市药物
MYC有所谓不可成药性,还没有专门的靶向药上市。
目前针对myc实际可行的治疗策略是根
与myc结合亲和力高的已上市药物
MYC有所谓不可成药性,还没有专门的靶向药上市。
目前针对myc实际可行的治疗策略是根






 显身卡
显身卡




